Python for Biologists
A collection of episodes with videos, codes, and exercises for learning the basics of the Python programming language through genomics examples.
Greetings! Thank you for joining us.
Here, you will find a series of episodes featuring examples from genomics to help you learn the basics of the Python programming language. No prior programming experience or knowledge of genetics is required.
Each episode includes a video and a working code highlighting a particular aspect of Python in the context of a genomics problem. The progression from episode to episode is nearly linear. After completing the final episode, you will be able to download a genome file from NCBI, and search and tally intricate motifs.
Mastering a programming language is best accomplished through active participation. As you watch each video, please code along with us, pausing as needed. It is critical to complete all exercises, as they enable you to assess your progress towards becoming an independent computational scientist.
In closing, we would like to express our gratitude to those who have contributed to both the conception and the realization of Python for Biologists. Colleagues Dimitris Papamichail and Burt Rosenberg, as well as former graduate assistants Andreas Seekircher and Justin Stoecker, helped with the development and testing of earlier versions of this material. Through their enthusiastic participation, our students aided us in fixing our ideas and setting realistic bounds for our own enthusiasm. In the final stages of the project, we benefitted from the assistance of Ziya Arnavut, Brian Coomes, Jason Glick, Denizen Kocak, Nancy Lawther, Joseph Masterjohn, Kenneth Palmer, Odelia Schwartz, and Stefan Wuchty.
Computational science is a flourishing frontier. It is our hope that Python for Biologists episodes will allow the subject of computational biology to come alive and tempt you to explore it further.
Hüseyin Koçak, Department of Computer Science, University of Miami
Basar Koc, Department of Computer Science, Stetson University
Introduction to Python
Welcome
# ------------------------------------------------------------------ # File name: welcome.py # # A line starting with # character is a comment and not interpreted # by Python. Use comments liberally in your codes. # # This is a customary first program to test if your computer is ready # for Python. Our program will simply print: # Welcome to Python for Biologists! # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # http://www.python.org # https://www.python.org/about/gettingstarted/ # ------------------------------------------------------------------ print('Welcome to Python for Biologists!')[csc210@boston myPython]$ python3 welcome.py Welcome to Python for Biologists!
Download

Beginnings
# ------------------------------------------------------------------- # File name: beginnings.py # # Variable names in Python start with a letter followed by # combination of letters, digits or underscore (no white spaces). # # Four of the basic variable types in Python are # numeric (integers and floats), string, list, and tuple. # The code below introduces examples of these variable types. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/introduction.html#an-informal-introduction-to-python # https://www.ncbi.nlm.nih.gov/nuccore/U00096 # https://en.wikipedia.org/wiki/Chargaff%27s_rules # ------------------------------------------------------------------ # String variable organism = "Escherichia coli" # NCBI accession number U00096 strain = 'str. K-12 substr. MG1665' print("DEFINITION: " + organism + " " + strain) # Integer variable number_of_bps = 4641652 print('Number of base pairs:', number_of_bps) # Float variable percent_A = 24.7 percent_T = 23.6 # List variable percents_AGCT = [percent_A, 26.0, 25.7, percent_T] print("[A, G, C, T] =", percents_AGCT) # Computing ratios A/T and G/C ratio_AT = percent_A / percent_T ratio_GC = percents_AGCT[1] / percents_AGCT[2] # Tuple variable E_Coli = (organism, ratio_AT, ratio_GC) print(E_Coli)[csc210@boston myPython]$ python3 beginnings.py DEFINITION: Escherichia coli str. K-12 substr. MG1665 Number of base pairs: 4641652 [A, G, C, T] = [24.7, 26.0, 25.7, 23.6] ('Escherichia coli', 1.0466101694915253, 1.0116731517509727)
Download

# ------------------------------------------------------------------ # File name: print.py # # print(*objects, sep=' ', end='\n', file=sys.stdout, flush=False) # The print() function writes the value of the argument(s) it is given. # It handles multiple arguments, floating point quantities, and strings. # Strings are printed without quotes, and a space is inserted between items. # The keyword argument end can be used to avoid the newline (\n) after the output # or end the output with a different string. # # You can escape (overrule its special meaning) a character by # prefixing it with backslash \ # # The code below illustrates some of the basic usages of the print() function. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/functions.html#print # https://docs.python.org/3/library/stdtypes.html#printf-style-string-formatting # https://en.wikipedia.org/wiki/ASCII # ------------------------------------------------------------------ import math human_genes = 20000 print('You have', human_genes, 'genes') print() # Replacing \n with a string print('You have', end =' ') print(human_genes, end = '? ') print('genes') # Spreading over lines print('You have', human_genes, 'genes') # Escaping with \ and string concatenation print('You have ' + '\'' + str(human_genes) + '\'' + ' genes') # printf style string formatting print('The value of pi is %10s %5.3f' %('--->', math.pi)) print(chr(7))[csc210@boston myPython]$ python3 print.py You have 20000 genes You have 20000? genes You have 20000 genes You have '20000' genes The value of pi is ---> 3.142
Download

Buggy
# ------------------------------------------------------------------ # File name: buggy.py # This short Python code contains a number of interntional bugs. Correct # them with the help of the error messages. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://americanhistory.si.edu/collections/search/object/nmah_334663 # https://en.wikipedia.org/wiki/Software_bug # ------------------------------------------------------------------ human genes = 20,000 protein-name = "GFP'; print('You have', human_genes, 'genes') print("protein-name stands for green fluorescent protein")[csc210@boston myPython]$ python3 buggy.py File "buggy.py", line 4 This short Python code contains a number of interntional bugs. Correct ^ IndentationError: unexpected indent
Download

- ASCII art:
Write a Python program that prints out the image below.
__ | \ |\ | /\ |__/ | \| /--\
Note: You can copy the image and paste it into your editor. To print the escape character \ you need to use \\ .
Personal challenge: Design your own letters to print out your initials. - Finding bugs:
Find and exterminate the bugs in the Python code below
# Please correct my errors. first_10_bp = "gggtgcgacg' second 10_bp = "attcattgtt" gene = first_10-bp + second 10_bp print('The first 20 bp of BRAC2 gene are gene']so that it prints out:The first 20 bp of BRAC2 gene are gggtgcgacgattcattgtt
- Pretty prints:
When precise control with the output of your program is needed
(lining up columns, decimal points, etc.), the
print function of Python
is your tool. It takes two arguments:
print("FORMAT" % (VALUES))FORMAT examples are: %15s field of width 15 for a string; %6d for an integer of width 6; %4.2f for a floating number with 4 digits to the left and 2 digits to the right of the decimal point.Now, using the statement
print("%15s %10s %10s" % ("Amino acid", "1-letter", "codon"))as a template, write a Python code that prints out the tableAmino acid 1-letter codon --------------- ---------- ---------- Serine S AGT AGC Cyteine C TGT
Data Types
Numeric
# ------------------------------------------------------------------- # File name: numeric.py # # Variable names in Python start with a letter followed by combination of # letters, digits or underscore (no white spaces). # # This program illustrates some of the basics of Python's numeric data types # int (for integer) and float. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/stdtypes.html#numeric-types-int-float-complex # https://docs.python.org/3/library/sys.html#sys.float_info # https://docs.python.org/3/library/math.html#module-math # https://docs.python.org/3/tutorial/controlflow.html#intermezzo-coding-style # https://www.census.gov/popclock/ # https://www.nature.com/articles/d41586-018-05462-w # https://en.wikipedia.org/wiki/Exon # https://en.wikipedia.org/wiki/Double-precision_floating-point_format # ------------------------------------------------------------------ import math # to use mathematical functions # Integers have unlimited precision human_genes = 21306 # int type. Not 21,306 or 21 306 US_population = 328918373 print('Number of human genes:', human_genes) print('Number of human genes in US:', human_genes*US_population) # Floats are implemented using double in C exons_per_gene = 8.9 # float type print('Human exons per gene:', exons_per_gene) human_exons = exons_per_gene*human_genes print('Number of human exons:', human_exons) # To convert float to integer human_exons = int(human_exons) print('Approximate number of human exons:', human_exons) # Can Python do arithmetic? firstProduct = (9.4*0.2321)*5.6 secondProduct = 9.4*(0.2321*5.6) print('(9.4*0.2321)*5.6 - 9.4*(0.2321*5.6) =', (firstProduct - secondProduct)) # To access mathematical functions from math module two_pi = 2.0*math.pi print('two_pi =', two_pi) print('sin(two_pi) =', math.sin(two_pi)) print('Do you believe this result?')[csc210@boston myPython]$ python3 numeric.py Number of human genes: 21306 Number of human genes in US: 7007934855138 Human exons per gene: 8.9 Number of human exons: 189623.4 Approximate number of human exons: 189623 (9.4*0.2321)*5.6 - 9.4*(0.2321*5.6) = -1.7763568394002505e-15 two_pi = 6.283185307179586 sin(two_pi) = -2.4492935982947064e-16 Do you believe this result?
Download

Strings
# ------------------------------------------------------------------- # File name: strings.py # # String variable names in Python start with a letter followed by # combination of letters, digits or underscore (no white spaces). # String literals are enclosed in single '...' or double "..." quotes. # Strings can be indexed, with the first character having index 0. # Indices may also be negative, to start counting from the right with -1. # Python strings cannot be changed — they are immutable. Therefore, # assigning to an indexed position in the string results in an error. # # The code below illustrates some of the basic string operations on a # partial DNA and protein sequences of GFP. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/introduction.html#strings # https://docs.python.org/3/library/stdtypes.html#common-sequence-operations # https://docs.python.org/3/library/stdtypes.html#text-sequence-type-str # https://docs.python.org/3/library/functions.html#len # https://en.wikipedia.org/wiki/Green_fluorescent_protein # https://www.nobelprize.org/prizes/chemistry/2008/summary/ # https://scienceblogs.com/pharyngula/2008/10/08/gfp-wins-nobel-prize # https://en.wikipedia.org/wiki/DNA_codon_table # ------------------------------------------------------------------ # String variables and literals protein = "GFP" # Winner of 2008 Nobel in chemistry protein_seq_begin = 'MSKGEELFTG' protein_seq_end = 'HGMDELYK' # Concatenation of strings protein_seq = protein_seq_begin + '...' + protein_seq_end print('Protein sequence of GFP: ' + protein_seq) # String method str.upper() DNA_seq = 'atgagtaaag...actatacaaa' DNA_seq = DNA_seq.upper() print('DNA sequence: ' + DNA_seq) # Forward index starts with 0 and increases # Backward index starts with -1 and decreases print('The second nucleotide:', DNA_seq[1]) print('The last nucleotide:', DNA_seq[-1]) # Slicing a string first_codon = DNA_seq[0:3] # index 3 excluded last_codon = DNA_seq[-3:] print('First codon:', first_codon) print('Last codon:', last_codon)[csc210@boston myPython]$ python3 strings.py Protein sequence of GFP: MSKGEELFTG...HGMDELYK DNA sequence: ATGAGTAAAG...ACTATACAAA The second nucleotide: T The last nucleotide: A First codon: ATG Last codon: AAA
Download

List
# ------------------------------------------------------------------- # File name: list.py # # Python compound data type list can be written as a list of # comma-separated values (items) between square brackets [,,]. Lists might # contain items of different types, but usually the items all have the # same type. Unlike strings, lists are a mutable type, i.e. it is # possible to change their content. Forward index starts with 0 and # increases; backward index starts with -1 and decreases. # # The code below illustrates some of the basic list operations using the # stop codons. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/introduction.html#lists # https://docs.python.org/3/tutorial/datastructures.html#more-on-lists # https://docs.python.org/3/library/stdtypes.html#lists # https://docs.python.org/3/library/stdtypes.html#common-sequence-operations # https://en.wikipedia.org/wiki/DNA_codon_table # ------------------------------------------------------------------ # Constructing a list stop_codons = ['TAA', 'tAG'] print(stop_codons) # Accessing an item in a list first_stop_codon = stop_codons[0] print(first_stop_codon) # Modifying an item in a list stop_codons[1] = 'TAG' print(stop_codons) # Appending an item to the end of a list stop_codons.append('TGA') print(stop_codons) # Number of items in a list number_of_stop_codons = len(stop_codons) print('There are', number_of_stop_codons, 'stop codons') # Convert list to a string DNA_seq = ''.join(stop_codons) print(DNA_seq) # Convert string to a list DNA_list = list(DNA_seq) print(DNA_list) # Slicing a list second_codon = DNA_list[3:6] # index 6 not included print('Second codon:', second_codon) # Copying a list DNA_list_duplicate = DNA_list.copy() print(DNA_list_duplicate) # Insert, delete element DNA_list_duplicate.insert(5, "?") print(DNA_list_duplicate) DNA_list_duplicate.pop(5) # Can also use: del DNA_list_duplicate[5] print(DNA_list_duplicate)[csc210@boston myPython]$ python3 list.py ['TAA', 'tAG'] TAA ['TAA', 'TAG'] ['TAA', 'TAG', 'TGA'] There are 3 stop codons TAATAGTGA ['T', 'A', 'A', 'T', 'A', 'G', 'T', 'G', 'A'] Second codon: ['T', 'A', 'G'] ['T', 'A', 'A', 'T', 'A', 'G', 'T', 'G', 'A'] ['T', 'A', 'A', 'T', 'A', '?', 'G', 'T', 'G', 'A'] ['T', 'A', 'A', 'T', 'A', 'G', 'T', 'G', 'A']
Download

Tuple
# ------------------------------------------------------------------- # File name: tuple.py # # Python compound data type tuple is an immutable sequence used to store # collections of heterogeneous or homogenous data. They can be constructed # in a number of ways; most typically, separating items with commas: # a, b, c or (a, b, c). Tuples implement all of the common sequence # operations. Some Python functions and methods return a list of tuples. # # The code below illustrates some of the common tuple operations using # basic amino acids and their codons. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/stdtypes.html#tuples # https://docs.python.org/3/library/stdtypes.html#common-sequence-operations # https://en.wikipedia.org/wiki/DNA_codon_table # ------------------------------------------------------------------ # Constructing tuples Histidine = ('H', 'CAT', 'CAC') Lysine = 'K', 'AAA', 'AAG' Arginine = ('R', 'CGT', 'CGC', 'CGA', 'CGG', 'AGA', 'AGG') print('Histidine:', Histidine) print('Lysine:', Lysine) print('Arginine:', Arginine) print() # Constructing a list of tuples basic = [Histidine, Lysine] basic.append(Arginine) print('Basic amino acids:', basic) print() # Accessing elements of a list of tuples His = basic[0] print('His:', His) His_codons = basic[0][1:] print('His codons:', His_codons) codon1, codon2 = His_codons print('codon1:', codon1) print('codon2:', codon2) print() protein_seq = basic[0][0] + basic[1][0] + basic[2][0] print('Protein:', protein_seq)[csc210@boston myPython]$ python3 tuple.py Histidine: ('H', 'CAT', 'CAC') Lysine: ('K', 'AAA', 'AAG') Arginine: ('R', 'CGT', 'CGC', 'CGA', 'CGG', 'AGA', 'AGG') Basic amino acids: [('H', 'CAT', 'CAC'), ('K', 'AAA', 'AAG'), ('R', 'CGT', 'CGC', 'CGA', 'CGG', 'AGA', 'AGG')] His: ('H', 'CAT', 'CAC') His codons: ('CAT', 'CAC') codon1: CAT codon2: CAC Protein: HKR
Download

Dictionary
# ------------------------------------------------------------------ # File name: dictionary.py # # A dictionary (dict) variable type is akin to a list that can hold # a number of values. However, instead of indexing with integers, it # uses a unique name, called a key, for each entry. A dict d = { } # can be defined by comma-separated pairs of key and value, with # a : in between, e.g. d = {"EcoRI" : "GAATTC"}. # # Code below illustrates some of the basic operations with a dictionary # containing several restriction enzymes and their recognition sites. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/datastructures.html#dictionaries # https://docs.python.org/3/library/stdtypes.html#mapping-types-dict # https://en.wikipedia.org/wiki/Restriction_enzyme # ------------------------------------------------------------------ restriction_enzymes = {'EcoRI' : 'GAATTC', 'AluI' : 'AGCT', 'NotI' : 'GCGGCCGC', 'TaqI' : 'TCGA' } print(restriction_enzymes) print() # To get a list of keys from a dictionary view object keys = list(restriction_enzymes.keys()) print('Keys as a list:', keys) # To get a list of values from a dictionary view object values = list(restriction_enzymes.values()) print('Values as a list:', values) # To check if a key is in the dictionary mykey = 'crispr' check = mykey in restriction_enzymes print('Is', mykey, 'key in the dictionary?', check) print() # To fetch a value from a dictionary with its key EcoRI_value = restriction_enzymes['EcoRI'] #raises a KeyError if key not found EcoRI_value = restriction_enzymes.get('EcoRI') # does not raise a KeyError if key not found print('The recognition site of EcoRI is', EcoRI_value) print() # To add to an existing dictionary restriction_enzymes['EcoRV'] = 'GATATC' restriction_enzymes.update(EcoRV = 'GATATC') print('With a new item:', restriction_enzymes) print() # To delete an item from a dictionary del restriction_enzymes['EcoRV'] print('Original dictionary:', restriction_enzymes) print()[csc210@boston myPython]$ python3 dictionary.py {'EcoRI': 'GAATTC', 'AluI': 'AGCT', 'NotI': 'GCGGCCGC', 'TaqI': 'TCGA'} Keys as a list: ['EcoRI', 'AluI', 'NotI', 'TaqI'] Values as a list: ['GAATTC', 'AGCT', 'GCGGCCGC', 'TCGA'] Is crispr key in the dictionary? False The recognition site of EcoRI is GAATTC With a new item: {'EcoRI': 'GAATTC', 'AluI': 'AGCT', 'NotI': 'GCGGCCGC', 'TaqI': 'TCGA', 'EcoRV': 'GATATC'} Original dictionary: {'EcoRI': 'GAATTC', 'AluI': 'AGCT', 'NotI': 'GCGGCCGC', 'TaqI': 'TCGA'}
Download

-
Arithmetic puzzle:
Create two numeric float variables
x = 0.1 * 10.0 y = 0.1 + 0.1 + 0.1 + 0.1 + 0.1 + 0.1 + 0.1 + 0.1 + 0.1 + 0.1
Print out the value ofx - y
Are you sure you did not make a mistake? LOL -
BRAC2 mRNA:
Visit the Web page
http://www.ncbi.nlm.nih.gov/nuccore/224004157?report=genbank
containing a partial coding mRNA sequence of the breast cancer gene BRAC2. You can read more about this gene at https://ghr.nlm.nih.gov/gene/BRCA2
Observe that the first 60 nucleotides of this gene are:gggtgcgacg attcattgtt ttcggacaag tggataggca accactaccg gtggattgtc
Create a Python program that stores this sequence without the blank spaces in a string and prints out the string in lower case and also in upper case letters. - List of codons for acidic acids: Create a list variable that holds the DNA codons for Glutamic acid. Next, add to this list the DNA codons that code for Aspartic acid. Your new list now should hold all the DNA codons for acidic acids. Print the codons for acidic acids, one per line. You will need to look up the necessary codons in a DNA codon table.
-
append() and pop():
Sometimes it is convenient to add to or subtract from a list without worrying about
indecies.
The
append() function adds an element(s) to the end of a list:
codon_list.append('GAT')Now, redo the previous problem using the append() function.The pop() function of Python pops and returns the last value of a list, shortening the list by one element:
last_element = codon_list.pop()
-
Sorting a list of strings:
Sorting entries of a list is a common task.
The sorted() function of Python
takes a list and returns the sorted list:
sorted_list = sorted(unsorted_list)
By default, the elements are sorted lexically in standard string comparison order.Modify your Python code in the previous problem so that your code prints out the codons sorted lexically.
-
Sorting(In-place) a list of numbers:
The sort() function of Python
can be made to sort in-place according to desired comparisons; you should consults the Python documentation for further
information.
To sort a list of numbers numerically, use
numbers = [21, 13, 0, -3.1, 21, 33, 2] numbers.sort()
Now, write a Python program to sort the unsorted list of numbers above, and print the sorted list. Experiment with or without the optional argument sort(reverse=True).
- Dictionary of codons for acidic acids: Create a dictionary variable using the RNA codons that codes for acidic amino acids as keys and the names of the acidic acids as values: (key, value) are (codon, amino acid). Extract the keys list of your dictionary and print it. Extract the values list of your dictionary and print it. You will need to look up for the necessary codons in a RNA codon table.
- Dictionary of codons for all amino acids: Create a dictionary variable using the RNA codons that codes for all amino acids as keys and the names of the amino acids as values: (key, value) are (UUU, Phenylalanine). There should be 64 items in your dictionary.
- Sorting a dictionary by keys or by values:
The following code illustrates how to sort a dictionary by keys or by values.
import operator mydictionary = {"c" : "2", "b" : "3", "a" : "4", "d" : "1" } mydictionary_sorted_by_keys = sorted(mydictionary.items(), key=operator.itemgetter(0)) mydictionary_sorted_by_values = sorted(mydictionary.items(), key=operator.itemgetter(1))Modify the Python code in the previous exercise to sort your dictionary of all codons and their amino acids by values. Your output should start as follows:GCC - Alanine GCU - Alanine GCA - Alanine GCG - Alanine UAG - Amber(STOP) CGU - Arginine ... (*) The key(s) of a given value may appear in a different order.
String Manipulations
Length
# ------------------------------------------------------------------ # File name: length.py # # Built-in function len(s) # Return the length (the number of items) of an object. The argument # may be a sequence (such as a string, bytes, tuple, list, or range) # or a collection (such as a dictionary, set, or frozen set). # # Code below computes the number of nucleotides in a DNA segment. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/functions.html#len # http://www.ncbi.nlm.nih.gov/nuccore/226377833?report=fasta # ------------------------------------------------------------------ zika_DNA = 'AGTTGTTGATCTGTGT' zika_DNA_length = len(zika_DNA) print('The first', zika_DNA_length, 'nucleotides', 'of Zika virus DNA are', zika_DNA)[csc210@boston myPython]$ python3 length.py The first 16 nucleotides of Zika virus DNA are AGTTGTTGATCTGTGT
Download

Concatenation
# ------------------------------------------------------------------ # File name: concatenation.py # # Binary operator + concatenates two strings. # # The code below concatenates the first four codons for GFP. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://en.wikipedia.org/wiki/RNA_codon_table # http://www.abcam.com/recombinant-a-victoria-gfp-protein-ab84191.html # ------------------------------------------------------------------ GFP_seq = 'MSKGEELFTG...HGMDELYK' print('Green fluorescent protein sequence:', GFP_seq) M_codon = 'AUG' S_codon = 'UCA' K_codon = 'AAA' G_codon = 'GGU' RNA_seq = M_codon RNA_seq = RNA_seq + S_codon print('RNA sequence:', RNA_seq) RNA_seq = RNA_seq + K_codon + G_codon print(RNA_seq, 'could code amino acid sequence MSKG')[csc210@boston myPython]$ python3 concatenation.py Green fluorescent protein sequence: MSKGEELFTG...HGMDELYK RNA sequence: AUGUCA AUGUCAAAAGGU could code amino acid sequence MSKG
Download

Find
# ------------------------------------------------------------------ # File name: find.py # # str.find(sub[, start[, end]]) # Return the lowest index in the string where substring sub is found # within the slice s[start:end]. Optional arguments start and end are # interpreted as in slice notation. Return -1 if sub is not found. # # str.rfind(sub[, start[, end]]) # Return the highest index in the string where substring sub is found, # such that sub is contained within s[start:end]. Optional arguments # start and endare interpreted as in slice notation. Return -1 on failure. # # str.index(sub[, start[, end]]) and str.rindex(sub[, start[, end]]) # are like find() and rfind() but raise ValueError when the substring # sub is not found. # # str.count(sub[, start[, end]]) # Return the number of non-overlapping occurrences of substring sub # in the range [start, end]. Optional arguments start and end are # interpreted as in slice notation. # # Code below compute the number and the locations of codon CAT in # a DNA segment. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/stdtypes.html#str.find # https://docs.python.org/3/library/stdtypes.html#str.rfind # https://docs.python.org/3/library/stdtypes.html#string-methods # https://en.wikipedia.org/wiki/D-loop # http://www.ncbi.nlm.nih.gov/nuccore/AF176731.1?report=fasta # ------------------------------------------------------------------ chimp = 'GTACCACCTAAGTACTGGCTCATTCATTACAACCGGTATGTACTTCGTACATTACTGCCAGTCACCATGA' print('Chimp D-loop:', chimp) codon = 'CAT' # Check if subtring is in string is_in = codon in chimp print('Is codon', codon, 'in chimp:', is_in) # .count() counts how many times sub appears in string how_many = chimp.count(codon) print('How many times', codon, 'appears in chimp:', how_many) # .find() returns the lowest index first_index = chimp.find(codon) print('First', codon, 'index: ', first_index) second_index = chimp.find(codon, first_index + len(codon)) print('Second', codon, 'index: ', second_index) # .find() returns -1 if cannot find sub in string third_index = chimp.find(codon, 27, 55) print('Third', codon, 'index: ', third_index) # .rfind() returns the highest index last_index = chimp.rfind(codon); print('Last', codon, 'index: ', last_index)[csc210@boston myPython]$ python3 find.py Chimp D-loop: GTACCACCTAAGTACTGGCTCATTCATTACAACCGGTATGTACTTCGTACATTACTGCCAGTCACCATGA Is codon CAT in chimp: True How many times CAT appears in chimp: 4 First CAT index: 20 Second CAT index: 24 Third CAT index: 49 Last CAT index: 65
Download

Slice
# ------------------------------------------------------------------- # File name: slice.py # # s[i:j:k] slice of s from i to j with step k # # The slice of s from i to j is defined as the sequence of items with # index k such that i <= k < j. If i or j is greater than len(s), use len(s). # If i is omitted or None, use 0. If j is omitted or None, use len(s). # If i is greater than or equal to j, the slice is empty. # # If i or j is negative, the index is relative to the end of sequence # s: len(s) + i or len(s) + j is substituted. But note that -0 is still 0. # # The code below illustrates the use of slicing in extraction of # subsequences from a DNA sequence. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/introduction.html#strings # https://docs.python.org/3/library/stdtypes.html#str.find # https://docs.python.org/3/library/stdtypes.html#common-sequence-operations # https://docs.python.org/3/library/stdtypes.html#text-sequence-type-str # https://en.wikipedia.org/wiki/DNA_codon_table # https://en.wikipedia.org/wiki/D-loop # https://www.ncbi.nlm.nih.gov/nuccore/X90314.1?report=fasta # ------------------------------------------------------------------ human = 'TTCTTTCATGGGGAAGCAGATTTGGGTACCACCCAAGTATTGACTTACCCATCAACAACCGCTATGTATT' print('Human D-loop:', human) mycodon = 'CAT' # Find the lowest index of mycodon in human index_mycodon = human.find(mycodon) print('First', mycodon, 'index:', index_mycodon) # Extract the first codon after mycodon first_codon = human[index_mycodon + 3: index_mycodon + 6] print('First codon after', mycodon, ':', first_codon) # Extract the second codon after mycodon second_codon = human[index_mycodon + 6: index_mycodon + 9] print('Second codon after', mycodon, ':', second_codon) # Negative starting point counts from the end of the string. next_to_last_codon = human[-6:-3] print('Next to last codon:', next_to_last_codon) # Omitted second entry in slicing indicates to the end of string last_codon = human[-3:] print('Last codon:', last_codon)[csc210@boston myPython]$ python3 slice.py Human D-loop: TTCTTTCATGGGGAAGCAGATTTGGGTACCACCCAAGTATTGACTTACCCATCAACAACCGCTATGTATT First CAT index: 6 First codon after CAT : GGG Second codon after CAT : GAA Next to last codon: TGT Last codon: ATT
Download

Translate
# ------------------------------------------------------------------ # File name: translate.py # # static str.maketrans(x[, y[, z]]) # This static method returns a translation table usable for str.translate(). # If there are two arguments, they must be strings of equal length, # and in the resulting dictionary, each character in x will be mapped # to the character at the same position in y. # If there is a third argument, it must be a string, whose characters # will be mapped to None in the result. # # The code below computes the complementary strand of a DNA sequence. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/stdtypes.html#str.maketrans # https://docs.python.org/3/library/stdtypes.html#str.translate # http://www.ncbi.nlm.nih.gov/nuccore/226377833?report=fasta # https://en.wikipedia.org/wiki/DNA # ------------------------------------------------------------------ zika_DNA = 'AGTTGTTGATCTGTGTGAGTCAG' print("Direct strand: 5' " + zika_DNA + " 3'") complements = zika_DNA.maketrans('acgtACGT', 'tgcaTGCA') complement_seq = zika_DNA.translate(complements) bonds = " "*25 + "|"*len(zika_DNA) print(bonds) print("Complementary strand: 3' " + complement_seq + " 5'")[csc210@boston myPython]$ python3 translate.py Direct strand: 5' AGTTGTTGATCTGTGTGAGTCAG 3' ||||||||||||||||||||||| Complementary strand: 3' TCAACAACTAGACACACTCAGTC 5'
Download
Reverse
# ------------------------------------------------------------------ # File name: reverse.py # # Reversing a DNA sequence is a common operation in genetics when dealing # with a complementary strand. String slicing operation string[::-1] # returns the desired result. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/stdtypes.html#common-sequence-operations # https://en.wikipedia.org/wiki/Primer_%28molecular_biology%29#/media/Fi # http://www.ncbi.nlm.nih.gov/nuccore/226377833?report=fasta # ------------------------------------------------------------------ zika_DNA = 'AATCCATGGTTTCT' print('Zika segment\t\t:', zika_DNA) reversed_zika_DNA = zika_DNA[::-1] print('Reversed zika segment\t:', reversed_zika_DNA)[csc210@boston myPython]$ python3 reverse.py Zika segment : AATCCATGGTTTCT Reversed zika segment : TCTTTGGTACCTAA
Download

Replace
# ------------------------------------------------------------------ # File name: replace.py # # str.replace(old, new[, count]) # Return a copy of the string with all occurrences of substring old # replaced by new. If the optional argument count is given, only the # first count occurrences are replaced. # # The code below removes spaces and digits in a segment of DNA in # GenBank format and highlights the start codon ATG. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/stdtypes.html#str.replace # http://www.ncbi.nlm.nih.gov/nuccore/226377833?report=genbank # ------------------------------------------------------------------ import re zika_DNA = '601 catgtgtgac gccaccatga gttatgagtg' print('Original Zika DNA\t\t:', zika_DNA) # Replace space with nothing one time zika_DNA = zika_DNA.replace(' ', '', 1) print('Replace space with nothing\t:', zika_DNA) # Replace all space characters with nothing zika_DNA = zika_DNA.replace(' ', '') print('Replace spaces with nothing\t:', zika_DNA) # Substitute all digits with nothing using regular expressions zika_DNA = re.sub(r'[1234567890]', '', zika_DNA) print('Replace numbers with nothing\t:', zika_DNA)[csc210@boston myPython]$ python3 replace.py Original Zika DNA : 601 catgtgtgac gccaccatga gttatgagtg Replace space with nothing : 601catgtgtgac gccaccatga gttatgagtg Replace spaces with nothing : 601catgtgtgacgccaccatgagttatgagtg Replace numbers with nothing : catgtgtgacgccaccatgagttatgagtg
Download

- Codons in a list: Create a Python program that stores a 15-base long DNA coding sequence in a string variable and then extracts all 5 codons comprising the DNA sequence and stores them in a list. The use of slicing is mandatory. Your program should print the sequence and, on a new line, the codons comprising it, separated by tabs.
- Fore-and-aft: Using slicing, extract and print out the first 3 and the last 3 codons
of the
BRAC2 gene segment below:
gggtgcgacg attcattgtt ttcggacaag tggataggca accactaccg gtggattgtc
- Finding motifs: Create a Python program that computes and prints the number of nucleotides
that separate the first and last appearance of the motif AGAG in lower case, upper case,
or combination, in the DNA sequence
GACGTCGCCAGAGAggcataTAACGATAtgacacagagagagcaGAGACAAGT
- Reverse complement: Create a Python program that computes the
reverse complement of the DNA sequence below and prints the sequence and its reverse complement:
GACGTCGCCAGAGAggcataTAACGATAtgacacagagagagcaGAGACAAGT
- DNA to RNA: Write a Python program that prints out the corresponding RNA sequence of a given DNA sequence. Make sure to handle sequences both in lowercase and uppercase (map t -> u, T -> U).
- Counting base pairs:
Write a Python code that prints out the percentages of the four nucleotides A, C, G, T in the
following segment of the breast cancer gene
BRAC2:
gggtgcgacg attcattgtt ttcggacaag tggataggca accactaccg gtggattgtc
-
Modifying strings:
In the Python programming language, string variables are immutable (cannot be changed) but list
variables are mutable.
However, it is possible to convert a string to a list, modify the list, and convert the modified list back to a string.
Write a Python code that takes the string
BRAC2_seq = "gggtgcgacgattcattgttttcggacaag"
and causes two mutations: replace the 4th "g" with an "a" and delete the 4th "t".
Conditionals and Loops
If Else
# ------------------------------------------------------------------ # File name: if_else.py # # The compound statement if_else has the syntax # # if EXPR: # statements # else: # statements # # If EXPR is True the first block of statements are executed, otherwise # the second block of statements following else are executed. # # Code below determines if the first codon in a DNA segment # is the start codon ATG or not and reports the result. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/controlflow.html?#if-statements # https://docs.python.org/3/reference/compound_stmts.html?#the-if-statement # https://docs.python.org/3/library/stdtypes.html?#truth-value-testing # ------------------------------------------------------------------ DNA_segment = 'ATGACATGA' codon1 = DNA_segment[0:3] # == operator tests the equality of two strings, resulting in True/False if (codon1 == 'ATG'): print('Codon', codon1, 'is a start codon.') else: print('Codon', codon1, 'is not a start codon.') print('Done!')[csc210@boston myPython]$ python3 if_else.py Codon ATG is a start codon. Done!
Download

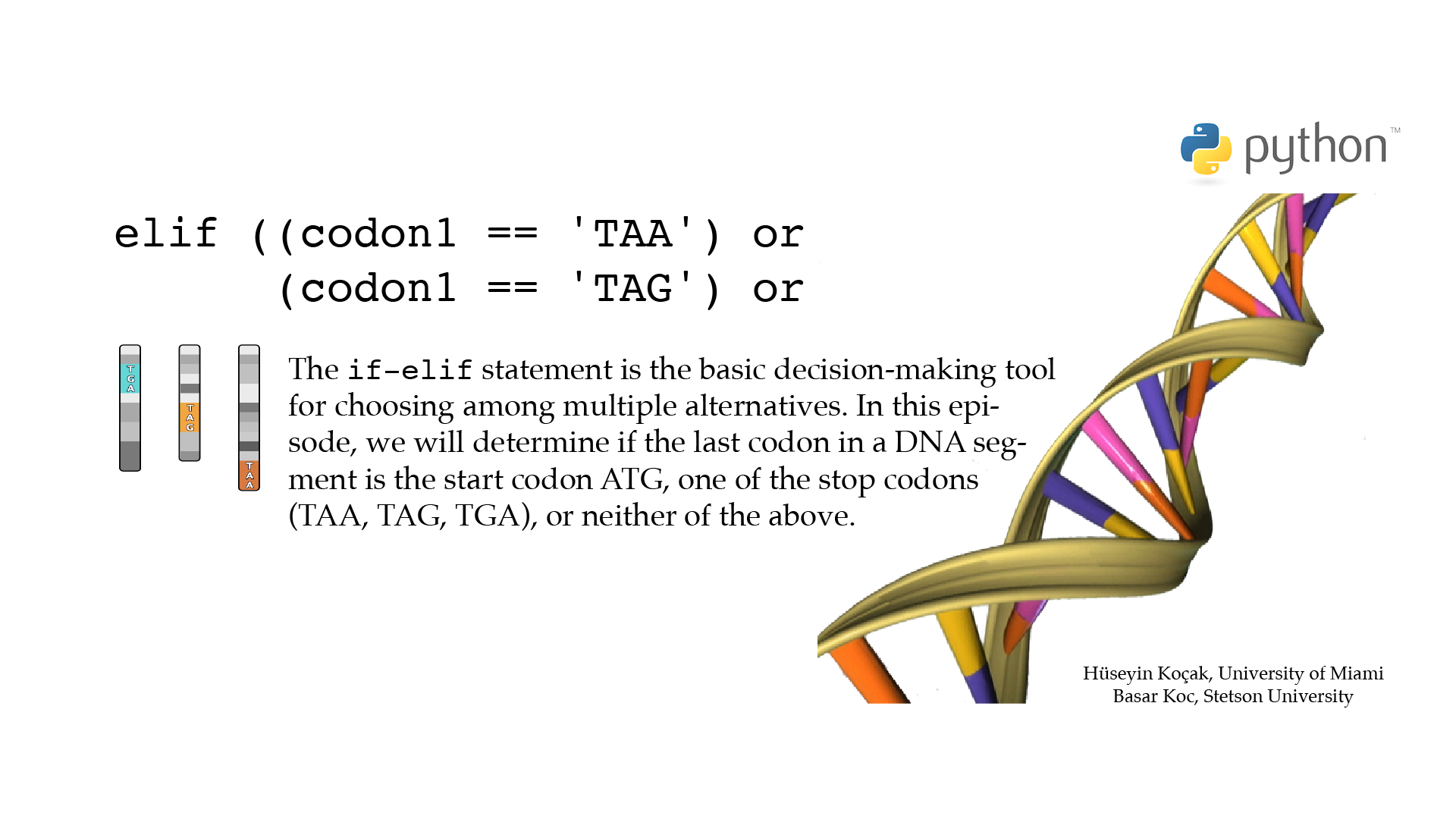
If Elif
# ------------------------------------------------------------------ # File name: if_elif.py # # The compound statement if_elif_else has the syntax # # if EXPR1: # statements1 # elif EXP2: # statements2 # else: # statements3 # # If EXPR1 is True, the first group of statements1 are executed and # the rest is skipped; otherwise, if EXPR2 is True, the statements2 # are executed; otherwise, statements3 are executed. # There can be zero or more elif parts, and the else part is optional. # The keyword ‘elif’ is short for ‘else if’, and is useful to avoid # excessive indentation. # An if … elif … elif … sequence is a substitute for the switch or # case statements found in other languages. # # Code below determines if the last codon in a DNA segment # is the start codon ATG or one of the stop codons TAA, TAG, or TGA; # or none of the above. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/controlflow.html?#if-statements # https://docs.python.org/3/reference/compound_stmts.html?#the-if-statement # https://docs.python.org/3/library/stdtypes.html?#truth-value-testing # https://docs.python.org/3/tutorial/datastructures.html?#comparing-sequences-and-other-types # https://en.wikipedia.org/wiki/DNA_codon_table # ------------------------------------------------------------------ DNA_segment = 'ATGACATGACCAATC' codon1 = DNA_segment[-3:] # == operator tests the equality of two strings, resulting in True/False if (codon1 == 'ATG'): print('Codon', codon1, 'is a start codon.') elif ((codon1 == 'TAA') or (codon1 == 'TAG') or (codon1 == 'TGA')): print('Codon', codon1, 'is a stop codon.') else: print('Codon', codon1, 'is neither a start nor a stop codon.') print('Done!')[csc210@boston myPython]$ python3 if_elif.py Codon ATC is neither a start nor a stop codon. Done!
Download
While
# ------------------------------------------------------------------ # File name: while.py # # while EXPR: # statements # # while loop iterates the block of statements as long as EXPR remains True. # # To illustrate the usage of while statement, the code below first # computes the number of appearances of a nucleotide base in a string # using Python's str.count() method. Then it computes the same number # using a while statement, hoping to get the same answer. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/reference/compound_stmts.html#the-while-statement # https://docs.python.org/3/library/stdtypes.html#string-methods # https://www.ncbi.nlm.nih.gov/nuccore/KC545393.1?report=fasta # ------------------------------------------------------------------ DNA_seq = 'CGGACACACAAAAAGAATGAAGGATTTTGAATCTTTATTGTGTGCGAGTAACTACGAGGAAGATTAAAGA' print('DNA sequence:', DNA_seq) bp = 'T' print('Base pair:', bp) print('str.count():', DNA_seq.count(bp)) count = 0 index = 0 while index < len(DNA_seq): if bp == DNA_seq[index]: count += 1 index += 1 print('Our while count:', count)[csc210@boston myPython]$ python3 while.py DNA sequence: CGGACACACAAAAAGAATGAAGGATTTTGAATCTTTATTGTGTGCGAGTAACTACGAGGAAGATTAAAGA Base pair: T str.count(): 17 Our while count: 17
Download

For
# ------------------------------------------------------------------ # File name: for.py # # for item in items: # statements # Python’s for statement iterates over the items of any sequence # (a list or a string), in the order that they appear in the sequence. # # Code below prints out all codons starting with T. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/controlflow.html#for-statements # https://docs.python.org/3/reference/compound_stmts.html#for # https://en.wikipedia.org/wiki/DNA_codon_table # ------------------------------------------------------------------ # Save nucleotide bases in a list bases = ['T', 'C', 'A', 'G'] # As a warmup, print the list of bases for base in bases: print(base) print('Codons starting with T:') for second_base in bases: print('Codons starting with T'+second_base) for third_base in bases: print('T'+second_base+third_base)[csc210@boston myPython]$ python3 for.py T C A G Codons starting with T: Codons starting with TT TTT TTC TTA TTG Codons starting with TC TCT TCC TCA TCG Codons starting with TA TAT TAC TAA TAG Codons starting with TG TGT TGC TGA TGG
Download

Range
# ------------------------------------------------------------------ # File name: range.py # # range(start, stop[, step]) # This built-in function generates arithmetic progressions. # The object returned by range() is not a list, but often acts like one. # The arguments to the range constructor must be integers. If the step # argument is omitted, it defaults to 1. If the start argument is omitted, # it defaults to 0. If step is zero, ValueError is raised. # For a positive step, the contents of a range r are determined by the # formula r[i] = start + step*i where i >= 0 and r[i] < stop. # # int() returns the integer part of a decimal number. # # Problem: Assume the population of Florida sandhill cranes grows by # 1.94% annually. If we start with a population of 425 birds, how large # will the population be after 30 years? Using a for loop, code below # computes and prints the population sizes for 30 years. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/controlflow.html#the-range-function # https://docs.python.org/3/library/stdtypes.html#ranges # https://sora.unm.edu/sites/default/files/journals/jfo/v061n02/p0224-p0231.pdf # https://www4.swfwmd.state.fl.us/springscoast/sandhillcranes.shtml # ------------------------------------------------------------------ # Initial population data population = [425] growth_rate = 0.0194 number_of_years = 30 # Construct a list numbering the years, from 0 to 30 years = range(0, number_of_years + 1, 1) #31 excluded print(list(years)) # Compute and append the population sizes to a list for year in years: next_generation = population[year] + growth_rate*population[year] population.append(next_generation) # Print the list of population sizes for year in years: print('At year %2d the population is %7.3f' %(year, population[year])) print() # Print the calculated population sizes rounded to the nearest integer for year in years: print('At year %2d the population is %3d' %(year, round(population[year])))[csc210@boston myPython]$ python3 range.py [0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30] At year 0 the population is 425.000 At year 1 the population is 433.245 At year 2 the population is 441.650 At year 3 the population is 450.218 At year 4 the population is 458.952 At year 5 the population is 467.856 At year 6 the population is 476.932 At year 7 the population is 486.185 At year 8 the population is 495.617 At year 9 the population is 505.232 At year 10 the population is 515.033 At year 11 the population is 525.025 At year 12 the population is 535.210 At year 13 the population is 545.593 At year 14 the population is 556.178 At year 15 the population is 566.968 At year 16 the population is 577.967 At year 17 the population is 589.179 At year 18 the population is 600.610 At year 19 the population is 612.261 At year 20 the population is 624.139 At year 21 the population is 636.248 At year 22 the population is 648.591 At year 23 the population is 661.173 At year 24 the population is 674.000 At year 25 the population is 687.076 At year 26 the population is 700.405 At year 27 the population is 713.993 At year 28 the population is 727.844 At year 29 the population is 741.965 At year 30 the population is 756.359 At year 0 the population is 425 At year 1 the population is 433 At year 2 the population is 442 At year 3 the population is 450 At year 4 the population is 459 At year 5 the population is 468 At year 6 the population is 477 At year 7 the population is 486 At year 8 the population is 496 At year 9 the population is 505 At year 10 the population is 515 At year 11 the population is 525 At year 12 the population is 535 At year 13 the population is 546 At year 14 the population is 556 At year 15 the population is 567 At year 16 the population is 578 At year 17 the population is 589 At year 18 the population is 601 At year 19 the population is 612 At year 20 the population is 624 At year 21 the population is 636 At year 22 the population is 649 At year 23 the population is 661 At year 24 the population is 674 At year 25 the population is 687 At year 26 the population is 700 At year 27 the population is 714 At year 28 the population is 728 At year 29 the population is 742 At year 30 the population is 756
Download

Count with Dictionary
# ------------------------------------------------------------------ # File name: count_with_dictionary.py # # Code below counts the number of each codon that appears in a DNA # segment using a dictionary. Keys are the present codons and the values # are their numbers. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/stdtypes.html#list # https://docs.python.org/3/library/stdtypes.html#mapping-types-dict # http://www.ncbi.nlm.nih.gov/nuccore/XM_002295694.2?report=fasta # http://www.opensourceshakespeare.org/stats/ # ------------------------------------------------------------------ DNA_seq = 'gggtgcgacgattcattgttttcggacaagtggataggcaaccactaccggtggattgtc' print('Sequence:', DNA_seq) # Count the number of codons DNA_length = len(DNA_seq) number_of_codons = int(DNA_length/3) print() # Put codons in a list codon_list = [] for i in range(number_of_codons): codon_list.append(DNA_seq[i*3:i*3 + 3]) print('List of codons:', codon_list) print() #Create codon counter dictionary (codon : codon_count) codon_counter = {} for codon in codon_list: if codon not in codon_counter: codon_counter[codon] = 1 else: codon_counter[codon] += 1 # This loop syntax accesses the whole dictionary by looping # over the .items() tuple list, accessing one (key, value) # pair at each step. print('Codon counter:') for key, value in codon_counter.items(): print(key, ':', value)[csc210@boston myPython]$ python3 count_with_dictionary.py Sequence: gggtgcgacgattcattgttttcggacaagtggataggcaaccactaccggtggattgtc List of codons: ['ggg', 'tgc', 'gac', 'gat', 'tca', 'ttg', 'ttt', 'tcg', 'gac', 'aag', 'tgg', 'ata', 'ggc', 'aac', 'cac', 'tac', 'cgg', 'tgg', 'att', 'gtc'] Codon counter: ggg : 1 tgc : 1 gac : 2 gat : 1 tca : 1 ttg : 1 ttt : 1 tcg : 1 aag : 1 tgg : 2 ata : 1 ggc : 1 aac : 1 cac : 1 tac : 1 cgg : 1 att : 1 gtc : 1
Download

- Searching for patterns:
Create a Python program that can check whether a given DNA sequence
contains the pattern
TATAxxxATGxxxT
where xxx can be any three consecutive DNA bases. That means your program, given some string, should identify if TATA, followed by any three DNA base pairs, followed by ATG, followed by any three DNA base pairs, followed by T can be found in the given string. Check both the forward and reverse complementary strand (reverse complement). Create three examples, one with a DNA string that contains the pattern in the forward strand, one that contains the pattern in the reverse complement strand and one that does not contain it in either strand, to demonstrate how the program works. Please include print statements that identify each different case and the position of the pattern in the string, if found. - Codons in array: Create a Python program that stores a DNA sequence of any length, extracts all codons in the sequence, and stores them in a list. Print the sequence and the codons, one per line, comprising it. Your program should handle the case where the sequence length is not a multiple of 3. Test your program with several sequences.
- Logistic growth with while and for with range:
Consider the discrete logistic model
x[n+1] = r * x[n] * (1.0 - x[n])
governing the growth of a single population with non-overlapping generations. Here, r is the growth rate and x[n] is the density of the n-th generation. Fix the growth rate and the initial population density asr = 3.73 and x[0] = 0.43.
Write a Python program that computes the population densities for 12 generations using a while statement, and prints out each generation and its population density on a separate line for all 12 generations.Now, write a second Python program that accomplishes the same task using a for statement with range.
- Locating substrings: Python code while.py computes the number of occurances of a nucleotide base in a DNA string. Modify this code to count the number of occurances of a given substring in a DNA string.
- While or for: Python code while.py computes the number of occurances of a nucleotide base in a DNA string. Replace the while loop with a for loop to achieve the same result.
- Timing: Determining the
running time of an algorithm is a key concern in computer science.
Python module
time
provides various time-related functions.
Be aware that "although this module is always available,
not all functions are available on all platforms."
Python code while.py in our Web site computes the number of occurrences of a nucleotide base in a DNA string with two different methods:
- Method 1: the built-in string method str.count()
- Method 2: a while loop.
- import time
- Run time.time() function immediately before and after each method to compute the running time of the method
- Print out the running time for each method and the ratio of the running time of method 2 over the running time of method 1
- Run your code several times. Do you get the same running times and ratios?
- In while.py the length of DNA_seq is 70. To get a longer sequence, you can make copies of this string with DNA_seq*N where N is a positive integer. Take N = 10, 100, 1000, 10000, and compute the running times and their ratios. How do they depend on the length of the string?
- Repeat the above steps using, this time, the function time.process_time()
- Summarize all your findings.
- All codons: Using as many for statements as necessary, write a Python program that prints out all 64 codons.
- Locating cutting sites of restriction enzymes:
In the episode on dictionaries, we created a dict varible with the names of restriction enzymes as keys
and the corresponding recognition sequences as values:
restriction_enzymes = {"EcoRI" : "GAATTC", "AluI" : "AGCT", "NotI" : "GCGGCCGC", "TaqI" : "TCGA" }Write a Python code to determine if the DNA sequenceDNAseq = "GGCGATGCTAGTCGCGTAGTCTAAGCTGTCGAGAATTCGGATGTCATGA"
will be cut with any of these restriction enzymes. Print out the location (index) of the recognition sites in the DNA sequence. For this problem, you should loop through the keys of the dict variable. - Generating random DNA sequences: Generate a random DNA string of 1001 bases
long and store it in a string variable. To do this, use a list of nucleotides and the
random.randint() function
from the random module of
Pyhon. Print out the percentages of the four nucleotides in your random DNA string.
Run your program several times. Do you get the same random sequence? Unlikely! For certain simulations, it may be necessary to use the same random sequence. Consult the documentation on how to set the seed of the random.seed() Python function.
- Counting k-mers:
Extract all substrings of length 4 (4-mers) from the sequence
DNA_seq = 'gggtgcgacgattcattgttttcggacaagtggataggcaaccactaccggtggattgtc'
and store them in a dictionary. Use the 4-mers as keys and the number of appearances as values in the dictionary. Print out all 4-mers in DNA_seq. - Generating random human DNA sequences: Chargaff rule states that "a double-stranded DNA molecule globally has percentage base pair equality: %A = %T and %G = %C." However, %A and %G varies with organisms. For instance, humans have, approximately, 29% A and 21% C. Write a Python program that generates random DNA sequences with percentages of A, T, C, and G of human DNA.
- Random mutations:
Write a Python program that causes three random mutations in the following DNA sequence
DNAseq = "GGCGATGCTAGTCGCGTAGTCTAAGCTGTCGAGAATTCGGATGTCATGA"
Make sure that both the locations and mutation types (replacement, insertion, and deletion) are random.
Utilities
Argv
# ------------------------------------------------------------------ # File name: argv.py # # Command line arguments are provided directly after the name of the # program and they are stored as a list in sys.argv. The first entry # sys.argv[0] is the script name (it is operating system dependent # whether this is a full pathname or not). # # Our program will print out the list of arguments; will exit if # no argument is provided. Example usage: args.py zika_DNA.fasta # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/interpreter.html#argument-passing # https://docs.python.org/3/library/sys.html # https://docs.python.org/3/using/cmdline.html#command-line-and-environment # ------------------------------------------------------------------ import sys if len(sys.argv) == 1: print('Please provide a command line argument!') sys.exit() print('sys.argv list:', sys.argv) print('The first argument:', sys.argv[0]) print('The second argument:', sys.argv[1])[csc210@boston myPython]$ python3 argv.py Please provide a command line argument! [csc210@boston myPython]$ python3 argv.py Zika.fasta sys.argv list: ['argv.py', 'Zika.fasta'] The first argument: argv.py The second argument: Zika.fasta
Download

Input
# ------------------------------------------------------------------ # File name: input.py # # input([prompt]) # If the prompt argument is present, it is written to standard output # without a trailing newline. The function then reads a line from input, # converts it to a string (stripping a trailing newline), and returns # that. When EOF is read, EOFError is raised. input() returns a string; # cast it if a number is expected. # # This program first prompts the user to type an NCBI sequence number # and then echos it. Second, it illustrates the casting of string # input with int() for use in arithmetical operations by calculating # the sum of two user-typed numbers. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/functions.html#input # ------------------------------------------------------------------ sequence_number = input('Please type an NCBI sequence number: ') print('Your sequence number is', sequence_number) # Input returns a string starting_index = input('Please type a starting index: ') ending_index = input('Please type an ending index: ') print('I will compute the number of bps in this region...') # Must cast string inputs to int or float for arithmetical operations number_of_bps = int(ending_index) - int(starting_index) print('The number of bps is:', number_of_bps)[csc210@boston myPython]$ python3 input.py Please type a NCBI sequence number: NC_012532.1 Your sequence number is NC_012532.1 Please type a starting index: 400 Please type an ending index: 1500 I will compute the number of bps in this region... The number of bps is: 1100
Download

Try/except
# ------------------------------------------------------------------ # File name: try_except.py # # Errors detected during execution are called exceptions. It is possible # to write programs that handle selected exceptions with a try-except statement. # The try statement works as follows: # 1. The statement(s) between the try and except keywords is executed. # 2. If no exception happens, the except block is skipped and execution # of the try statement is finished. # 3. If an exception occurs during execution of the try clause, the rest # of the clause is skipped. Then if its type matches the exception # named after the except keyword, the except clause is executed, and # then execution continues after the try statement. # 4. An unhandled exception stops the execution. # 5. Optional else clause must follow all except clauses. It is useful # for code that must be executed if the try clause does not raise # an exception. # # The code below illustrates the usage of try-except statement in handling # exceptions, e.g. input error (ValueError), out-of-bound index error # (IndexError) in a list. The program asks for an input, repeatedly, if an # exception is raised. If no exception is raised, the program breaks out of the # infinite while loop by executing the else clause and finishes with # a message. One can also exit the program by typing Control^C. # # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/errors.html#exceptions # https://docs.python.org/3/reference/compound_stmts.html#the-try-statement # https://docs.python.org/3/library/exceptions.html # ------------------------------------------------------------------ import sys stop_codons = ['TAA', 'TAG', 'TGA'] print('Stop codons:', stop_codons) while True: try: index = int(input('Please enter the index of a stop codon to print: ')) print('Your codon is', stop_codons[index]) except ValueError as ve: print(ve, 'Try again...') except IndexError: print('Your index', index, 'is out of range. Try again...') except: print('Unexpected error:', sys.exc_info()[0]) sys.exit() else: print('Good bye!') break[csc210@boston myPython]$ python3 try_except.py Stop codons: ['TAA', 'TAG', 'TGA'] Please enter the index of a stop codon to print: 1 Your codon is TAG Good bye!
Download

Function
# ------------------------------------------------------------------ # File name: function.py # # The keyword def introduces a function definition. It must be followed # by the function name and the parenthesized list of formal parameters. # The statements that form the body of the function start at the next line, # and must be indented. Variables in a function are local to that function. # # The first statement of the function body can optionally be a string literal # enclosed in triple quotes ''' ... '''; this string literal is the # function’s documentation string, or docstring. # # The user-defined function below takes a DNA segment as a string # and returns the corresponding RNA segment as a string. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/controlflow.html#defining-functions # https://docs.python.org/3/reference/compound_stmts.html#function-definitions # https://docs.python.org/3/tutorial/controlflow.html#documentation-strings # http://www.ncbi.nlm.nih.gov/nuccore/226377833?report=fasta # ------------------------------------------------------------------ def DNAtoRNA(dna): ''' Converts DNA string to RNA string. Parameters: DNA sequence as a string Return: RNA sequence as a string ''' transliterate = dna.maketrans('tT', 'uU') rna = dna.translate(transliterate) return rna # Can access the doctring ''' ... ''' with print(DNAtoRNA.__doc__) zika_DNA = 'AGTTGTTGATCTGTGTGAGTCAGACTGCG' print('Zika DNA segment is', zika_DNA) zika_RNA = DNAtoRNA(zika_DNA); print('Zika RNA segment is', zika_RNA)[csc210@boston myPython]$ python3 function.py Zika DNA segment is AGTTGTTGATCTGTGTGAGTCAGACTGCG Zika RNA segment is AGUUGUUGAUCUGUGUGAGUCAGACUGCG
Download

- Keyboard inputs: Write a Python program that asks the user to enter a DNA sequence from the keyboard and captures the sequence in a variable. Next, your program should ask the user for a second shorter DNA sequence to be entered. Now, your program should report if the second sequence is a substring of the first string.
- Arguments as input: Write a Python program that takes two strings as command-line arguments and reports if the second argument is a substring of the first argument.
- What does it do?
Consult the documentation on how to use the
sys.stdin() Python function. What does the Python code below do?
import sys text = "" while 1: c = sys.stdin.read(1) text = text + c if c == '\n': break print("Input: %s" % text) - Where is my number?
Modify the input.py code to catch exceptions if the user-entered numbers cannot be
converted to integers. Test your code with the inputs below:
Please type a codon: ATG Your codon is ATG Please type a starting index: 400 Please type an ending index: 1,500 I will compute the number of bps in this region... Traceback (most recent call last): File "input.py", line 11, in
number_of_bps = int(ending_index) - int(starting_index) ValueError: invalid literal for int() with base 10: '1,500' - A function returning a random sequence: Write a Python function that takes as a parameter the length and returns a random DNA sequence with the requested length.
- A function for growth of Florida sandhill cranes: In the range episode, we encountered the following problem: The population of Florida sandhill cranes in the Okefenokee swamp, under the current environmental conditions, grows by 1.94% annually. If we start with a population of 425 birds, how big will the population be after 28 years? Now, create a Python function that, given an initial population size, growth rate, and a number of years, can calculate and return the Florida sandhill crane population size after that time period.
- Modules:
Python has a way to put function definitions in a file and use them in a script. Such a file is called a module.
Definitions from a module can be imported into other modules or into the main program.
Consult the Python tutorial Modules for further
information.
Now, create a module named dna_rna.py that includes two function definitions DNAtoRNA() and RNAtoDNA(). Write a Python program to do the following:
1. Import the dna_rna module, 2. Take a DNA segment, 3. Convert to RNA segment using DNAtoRNA(), 4. Convert the return string back to DNA using RNAtoDNA(), 5. Check if you get the original DNA segment back.
- Percentages: Write a Python function that takes a DNA segment as a string and returns the percentages of A,C,G,T as a list. Make sure that your function handles both upper and lowercases.
- Reverse complement: Write a Python function that takes a DNA sequence as a string and returns its reverse complement.
- Two parameters: Write a Python function that takes a DNA sequence as a string and a nucleotide and returns the number of occurrences of the nucleotide in the sequence.
- Documentation Strings: A function's documentation string, or docstring, contains information about the function, its usage, etc. Consult here on guidelines for writing documentations strings. Now, write a docstring for your function in the previous problem.
Files
Write to File
# ----------------------------------------------------------------- # File name: write_to_file.py # # open(file, mode='r', buffering=-1, encoding=None, errors=None, # newline=None, closefd=True, opener=None) # This function opens file and returns a corresponding file object. # If the file cannot be opened, an OSError is raised. Once opened, # you can write to file with myfile.write() or print('string', file = myfile) # It is advantageous to use the with keyword when dealing with file # objects because the file is properly closed after its suite finishes, # even if an exception is raised at some point. # # Code below opens a file and writes information about a DNA sequence # in FASTA format. At the end, it reports if the file is created. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # http://www.esa.int/Our_Activities/Space_Science/Rosetta/Science_on_the_surface_of_a_comet # https://en.wikipedia.org/wiki/DNA_codon_table # https://docs.python.org/3/tutorial/inputoutput.html#reading-and-writing-files # https://docs.python.org/3/reference/compound_stmts.html#the-with-statement # https://docs.python.org/3/library/functions.html#open # https://docs.python.org/3/library/functions.html#print # ---------------------------------------------------------------- import os.path filename = 'Rosetta_partial.fasta' with open(filename, 'w') as f: print('>JB_007 Rosetta partial genome', file=f) f.write('ATG') f.write('GGT') f.write('GGC') f.write('GGA') f.write('GGG') f.write('TGA') f.write('xxxACCATG') f.write('\n') if os.path.isfile(filename): print('Rosetta partial genome is written to', filename, 'file successfully!')[csc210@boston myPython]$ python3 write_to_file.py Rosetta partial genome is written to Rosetta_partial.fasta file successfully!
Download

Read from File
# ------------------------------------------------------------------- # File name: read_from_file.py # # open(file, mode='r', buffering=-1, encoding=None, errors=None, # newline=None, closefd=True, opener=None) # This function opens file and returns a corresponding file object. # If the file cannot be opened, an OSError is raised. Once opened, # you can write to file with myfile.write() or print('string', file = myfile) # It is advantageous to use the with keyword when dealing with file # objects because the file is properly closed after its suite finishes, # even if an exception is raised at some point. If you are not using the # with keyword, then you should call myfile.close() to close the file. # # This program opens a file and demonsrates several ways of reading it. # The filename will be provided as a command line argument. This means # that it will be supplied when we run our program, directly after # the name of the program. Example: read_fasta_file.py BRAC2.fasta # Our program will exit if the file name is not provided, or the file # does not exist or cannot be opened. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/inputoutput.html#reading-and-writing-files # https://docs.python.org/3/library/functions.html#open # ------------------------------------------------------------------ import sys if len(sys.argv) == 1: print('Please provide a command line argument as a file name!') sys.exit() else: myfile = sys.argv[1] # Open the file for reading, and read the first five lines. Close file. try: f = open(myfile, 'r') except OSError as oserr: print('OS error:', oserr) else: print(f.readline(), end = '') print(f.readline(), end = '') print(f.readline(), end = '') print(f.readline(), end = '') f.close() print() # Read all lines into a list try: f = open(myfile, 'r') except OSError as oserr: print('OS error:', oserr) else: lines = f.readlines() number_of_lines = len(lines) print('There are', number_of_lines, 'lines in', myfile) f.close() print() # The best way to read all the lines of a file try: with open(myfile, 'r') as f: for line in f: print(line, end='') except OSError as oserr: print('OS error:', oserr)[csc210@boston myPython]$ python3 read_from_file.py Please provide a command line argument as a file name! [csc210@boston myPython]$ python3 read_from_file.py BRAC2.fasta >gi|224004157|ref|XM_002295694.1| Thalassiosira pseudonana CCMP1335 chromosome 7 breast cancer 2 early onset (BRAC2) mRNA, partial cds GGGTGCGACGATTCATTGTTTTCGGACAAGTGGATAGGCAACCACTACCGGTGGATTGTCTGGAAGCTAG CAGCAATGGAGAGACGGTTTCCACACCATCTTGGAGGACATTACTTGACGTACGAGCGTGTGCTGAAACA AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAGACGGCCTGCAGTACGCATAATGCTCAACCGA There are 16 lines in BRAC2.fasta >gi|224004157|ref|XM_002295694.1| Thalassiosira pseudonana CCMP1335 chromosome 7 breast cancer 2 early onset (BRAC2) mRNA, partial cds GGGTGCGACGATTCATTGTTTTCGGACAAGTGGATAGGCAACCACTACCGGTGGATTGTCTGGAAGCTAG CAGCAATGGAGAGACGGTTTCCACACCATCTTGGAGGACATTACTTGACGTACGAGCGTGTGCTGAAACA AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAGACGGCCTGCAGTACGCATAATGCTCAACCGA GATGTTGCAGCGAGTTTGCCAGTCATCTTATGCGTAAGCCAAATCCTTCGATTCAAATCAAGACCGCCAA AAGGAAGTTCTTCCGACGAGATCAAAGAAGAAGTCCGACTGGAGTTGACGGATGGATGGTACTCACTACC TGCTGTAGTGGACGAAATACTGTTGAAGTTTGTTGAAGAAAGGAGAATCGCAGTGGGATCAAAACTAATG ATTTGCAATGGGCAGTTAGTTGGATCTGATGACGGAGTGGAGCCTCTCGATGACAGCTACTCATCTTCCA AACGAGATTGTCCTCTATTGCTGGGCATCTCTGCCAACAACTCCCGTTTAGCAAGATGGGATGCAACTCT AGGTTTTGTACCTCGCAACAACTCTAATCTATACGGCGGCAATCTTTTGGTCAAATCCCTGCAAGACATT TTCATCGGCGGAGGTACTGTTCCGGCTATTGATTTGGTTGTTTGTAAGAAGTACCCAAGGATGTTTCTAG AGCAATTAAACGGTGGAGCTTCCATTCATCTTACAGAAGCCGAAGAAGCAGCACGCCAAAGTGAGTACGA TTCAAGGCATCAGCGAGCAAGCGAGAGATATGCCGACGATGCTACGAAGGAATGTTCAGAGGTAAGTTCA TTGCTGTTCACATTCTTCACTATGAAGCCACTTCCGTTGCTTTGGTACAATCTTGTCACTGACTCATCTT TTGGCGTTCATGATTCGCACAGGAAATCGATGAGGATGCTCCTACTCAGTGGAAAGAGATG
Download

Read FASTA File
# ------------------------------------------------------------------- # File name: read_fasta_file.py # # open(file, mode='r', buffering=-1, encoding=None, errors=None, # newline=None, closefd=True, opener=None) # This function opens file and returns a corresponding file object. # If the file cannot be opened, an OSError is raised. Once opened, # you can write to file with myfile.write() or print('string', file = myfile) # It is advantageous to use the with keyword when dealing with file # objects because the file is properly closed after its suite finishes, # even if an exception is raised at some point. # # Below, we will open a file containing information about a DNA sequence # in FASTA format. Then we will read the file line-by-line, except # line starting with > sign; remove newline characters and concatenate # lines into a single string. # # The filename will be provided as a command line argument. This means # that it will be supplied when we run our program, directly after # the name of the program. Example: read_fasta_file.py BRAC2.fasta # Our program will exit if the file name is not provided, or the file # does not exist or cannot be opened. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/tutorial/inputoutput.html#reading-and-writing-files # https://docs.python.org/3/library/functions.html#open # https://docs.python.org/3/library/stdtypes.html#str.strip # https://en.wikipedia.org/wiki/BRCA2 # http://www.ncbi.nlm.nih.gov/nuccore/XM_002295694.2?report=fasta # ------------------------------------------------------------------ import sys if len(sys.argv) == 1: print('Please provide a command line argument as a file name!') sys.exit() else: myfile = sys.argv[1] sequence = '' try: with open(myfile, 'r') as f: for line in f: if '>' not in line: sequence = sequence + line.strip() except OSError as oserr: print('OS error:', oserr) else: print(sequence)[csc210@boston myPython]$ python3 read_fasta_file.py Please provide a command line argument as a file name! [csc210@boston myPython]$ python3 read_fasta_file.py BRAC2.fasta GGGTGCGACGATTCATTGTTTTCGGACAAGTGGATAGGCAACCACTACCGGTGGATTGTCTGGAAGCTAGCAGCAATGGAGAGACGGTTTCCACACCATCTTGGAGGACATTACTTGACGTACGAGCGTGTGCTGAAACAAATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAGACGGCCTGCAGTACGCATAATGCTCAACCGAGATGTTGCAGCGAGTTTGCCAGTCATCTTATGCGTAAGCCAAATCCTTCGATTCAAATCAAGACCGCCAAAAGGAAGTTCTTCCGACGAGATCAAAGAAGAAGTCCGACTGGAGTTGACGGATGGATGGTACTCACTACCTGCTGTAGTGGACGAAATACTGTTGAAGTTTGTTGAAGAAAGGAGAATCGCAGTGGGATCAAAACTAATGATTTGCAATGGGCAGTTAGTTGGATCTGATGACGGAGTGGAGCCTCTCGATGACAGCTACTCATCTTCCAAACGAGATTGTCCTCTATTGCTGGGCATCTCTGCCAACAACTCCCGTTTAGCAAGATGGGATGCAACTCTAGGTTTTGTACCTCGCAACAACTCTAATCTATACGGCGGCAATCTTTTGGTCAAATCCCTGCAAGACATTTTCATCGGCGGAGGTACTGTTCCGGCTATTGATTTGGTTGTTTGTAAGAAGTACCCAAGGATGTTTCTAGAGCAATTAAACGGTGGAGCTTCCATTCATCTTACAGAAGCCGAAGAAGCAGCACGCCAAAGTGAGTACGATTCAAGGCATCAGCGAGCAAGCGAGAGATATGCCGACGATGCTACGAAGGAATGTTCAGAGGTAAGTTCATTGCTGTTCACATTCTTCACTATGAAGCCACTTCCGTTGCTTTGGTACAATCTTGTCACTGACTCATCTTTTGGCGTTCATGATTCGCACAGGAAATCGATGAGGATGCTCCTACTCAGTGGAAAGAGATG
Download

-
Counting Severe Acute Respiratory Syndrome coronavirus 2 genome bps:
Download the FASTA file (NC_045512.2) containing the SARS-CoV-2 reference sequence. (You can use the "Send" widget on the upper-right corner of
the NCBI Web page containing the genome to download a FASTA file.)
Now, write a Python program that reads the file, stores the sequence without white
characters (spaces, end-of-line, etc.),
and prints out the number of nucleotides (bps) in the complete SARS-CoV-2 genome.
- Start codons in the SARS-CoV-2 genome:
Modify your Python code in the previous problem so that your code
prints out the locations of the first appearances of the start codon ATG and one
of the stop codons (TAA, TAG, TGA) afterwords in the SARS-CoV-2 genome.
- SARS-CoV-2 genomic variations: Samples of this virus from various geographical locations display variations in the
genomic sequences. There are several hundred such sequences at GenBank, as linked in the References.
Here are four examples:
Wuhan-Hu-1: NC_045512.2
(considered the Reference)
India: MT050493.1
Italy: MT066156.1
U.S.A.: MN985325.1
- Download the sequences Wuhan-Hu-1 and U.S.A in FASTA format.
- Write a Python program that reads these files and saves the sequences as strings.
- Your program should compare the nucleotide sequences and print out the locations (indecies) where they differ and the differences. Note that these sequences are of different lengths; compare them only upto the length of the shorter one.
- BLAST (Basic Local Alignment Search Tool):
"BLAST
finds regions of similarity between biological sequences.
The program compares nucleotide or protein sequences to sequence
databases and calculates the statistical significance."
- Visit the BLAST Web site linked above and choose the icon for "Nucleotide BLAST."
- In the "Enter query Sequence" box enter one of the SARS-CoV-2`accession numbers from the list Wuhan-Hu-1: NC_045512.2 (considered the Reference) India: MT050493.1 Italy: MT066156.1 U.S.A.: MN985325.1
- Check "Align two or more sequences."
- In the newly opened "Enter Subject Sequence" box, another SARS-CoV-2`accession numbers from the list.
- Hit the "BLAST" button at the bottom of the page.
- Select "Alignments" option to see the comparison of the two sequences.
- Select for "Alignment view", the option "Pairwise with dots for identities", scroll down and looks for the differences in the two sequences.
- Report the differences in the genomic sequences.
- Logistic growth model:
Consider the discrete logistic model
x[n+1] = r * x[n] * (1.0 - x[n])
governing the growth of a single population with non-overlapping generations. Here, r is the growth rate and x[n] is the density of the n-th generation. Fix the growth rate r = 3.1 and initial population density x[0] = 0.43. Write a Python program that computes the population densities for 12 generations and writes them out to a file, each generation and its population density on a separate line, as below:At year 0, the population density is 0.43 At year 1, the population density is ...
- Copying a file:
Write a Python program that takes two file names as a command line argument
and copies the first FASTA file into the second file. Now, use the Unix command diff to test the output of your program.
-
Comparing k-mers in viral genomes:
Create a Python program to do the following:
- Open a FASTA file whose name is provided as a command line argument, concatenate the sequence lines in a string.
- Extract all substrings of length 9 (9-mers) from the sequence and store all 9-mers in a dictionary, together with the number of times they appear in the string. Use the 9-mers as keys and the number of appearances as values in the dictionary. At the end, the program should print all 9-mers and their counts.
- Now, edit the previous program (or create a new one) that opens and processes two separate virus genomes in FASTA format. Your goal is to compare the two genomes and determine the number of substrings of length 9 (9-mers) that they share. Please print all 9-mers that the two genomes share and their total number (count). Select two random genomes, preferably not longer than 10000 nucleotides each. You should supply the FASTA files with the virus genome sequences as command-line arguments to your program and use the sys.argv list to import the sequences.
- The two virus genomes can be downloaded from NCBI. For a starting point, you can use this webpage. Please include your choices virus names and accession numbers in your deliverable.
- Would you expect to get similar results if these were not virus genome sequences but random DNA/RNA sequences?
Regular Expressions
Motif Search
# ------------------------------------------------------------------ # File name: motif_search.py # # re — Regular expression operations # This module provides regular expression matching operations similar # to those found in Perl. # # re.search(pattern, string, flags=0) # Scan through string looking for the first location where the # regular expression pattern produces a match, and return a # corresponding match object. Return None if no position in the # string matches the pattern. # # This program reports if a motif (ATG followed by zero or more any # characters-non greedy-ending with TAA) is present in a DNA sequence, # and prints the matched substring, the start and end indices. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/re.html#module-re # https://docs.python.org/3/library/re.html#raw-string-notation # https://docs.python.org/3/library/re.html#re.search # https://docs.python.org/3/library/re.html#match-objects # https://docs.python.org/3/library/re.html#re.compile # http://www.ncbi.nlm.nih.gov/nuccore/XM_002295694.2?report=fasta # ------------------------------------------------------------------ import re import sys DNA_sequence = 'AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG' print('DNA_sequence:', DNA_sequence) motif = r'ATG.*?TAA' # r for raw string #motif = r'[ATG.*?TAA' # This is an invalid regular expression print('Motif:', motif) # Checking if motif is a valid regular expression try: re.compile(motif) except: print('Invalid regular expression, exiting the program!') sys.exit() match = re.search(motif, DNA_sequence) if match: print('Found the motif :', match.group()) print('Starting at index :', match.start()) print('Ending at index :', match.end()) else: print('Did not find the motif.')[csc210@boston myPython]$ python3 motif_search.py DNA_sequence: AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG Motif: ATG.*?TAA Found the motif : ATGAAGGGCCGCTACGATAA Starting at index : 1 Ending at index : 21
Download

Motif Search Groups
# ------------------------------------------------------------------ # File name: motif_search_groups.py # # Match.group([group1, ...]) # Returns one or more subgroups of the match. If there is a single argument, # the result is a single string; if there are multiple arguments, the result # is a tuple with one item per argument. Without arguments, group1 defaults # to zero (the whole match is returned). If a groupN argument is zero, the # corresponding return value is the entire matching string; if it is in the # inclusive range [1..99], it is the string matching the corresponding # parenthesized (...) group. # # This program reports if a motif (ATG followed by zero or more any # characters-non greedy-ending with TAA) is present in a DNA sequence # and lists the groupings (two groups are the motif found and the characters # between ATG and TAA in the found motif). # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/re.html#re.Match.group # https://docs.python.org/3/library/re.html#module-re # https://docs.python.org/3/library/re.html#raw-string-notation # https://docs.python.org/3/library/re.html#re.search # https://docs.python.org/3/library/re.html#match-objects # http://www.ncbi.nlm.nih.gov/nuccore/XM_002295694.2?report=fasta # ------------------------------------------------------------------ import re import sys DNA_sequence = 'AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG' print('DNA_sequence:', DNA_sequence) motif = r'(ATG(.*?)TAA)' print('Motif:', motif) # Checking if motif is a valid regular expression try: re.compile(motif) except: print('Invalid regular expression, exiting the program!') sys.exit() match = re.search(motif, DNA_sequence) if match: print('group0 :', match.group(0)) print('group0 start-end :', match.start(0), match.end(0)) print('group1 :', match.group(1)) print('group1 start-end :', match.start(1), match.end(1)) print('group2 :', match.group(2)) print('group2 start-end :', match.start(2), match.end(2)) print('groups as tuples :', match.groups()) else: print('Did not find the motif.')[csc210@boston myPython]$ python3 motif_search_groups.py DNA_sequence: AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG Motif: (ATG(.*?)TAA) group0 : ATGAAGGGCCGCTACGATAA group0 start-end : 1 21 group1 : ATGAAGGGCCGCTACGATAA group1 start-end : 1 21 group2 : AAGGGCCGCTACGA group2 start-end : 4 18 groups as tuples : ('ATGAAGGGCCGCTACGATAA', 'AAGGGCCGCTACGA')
Download
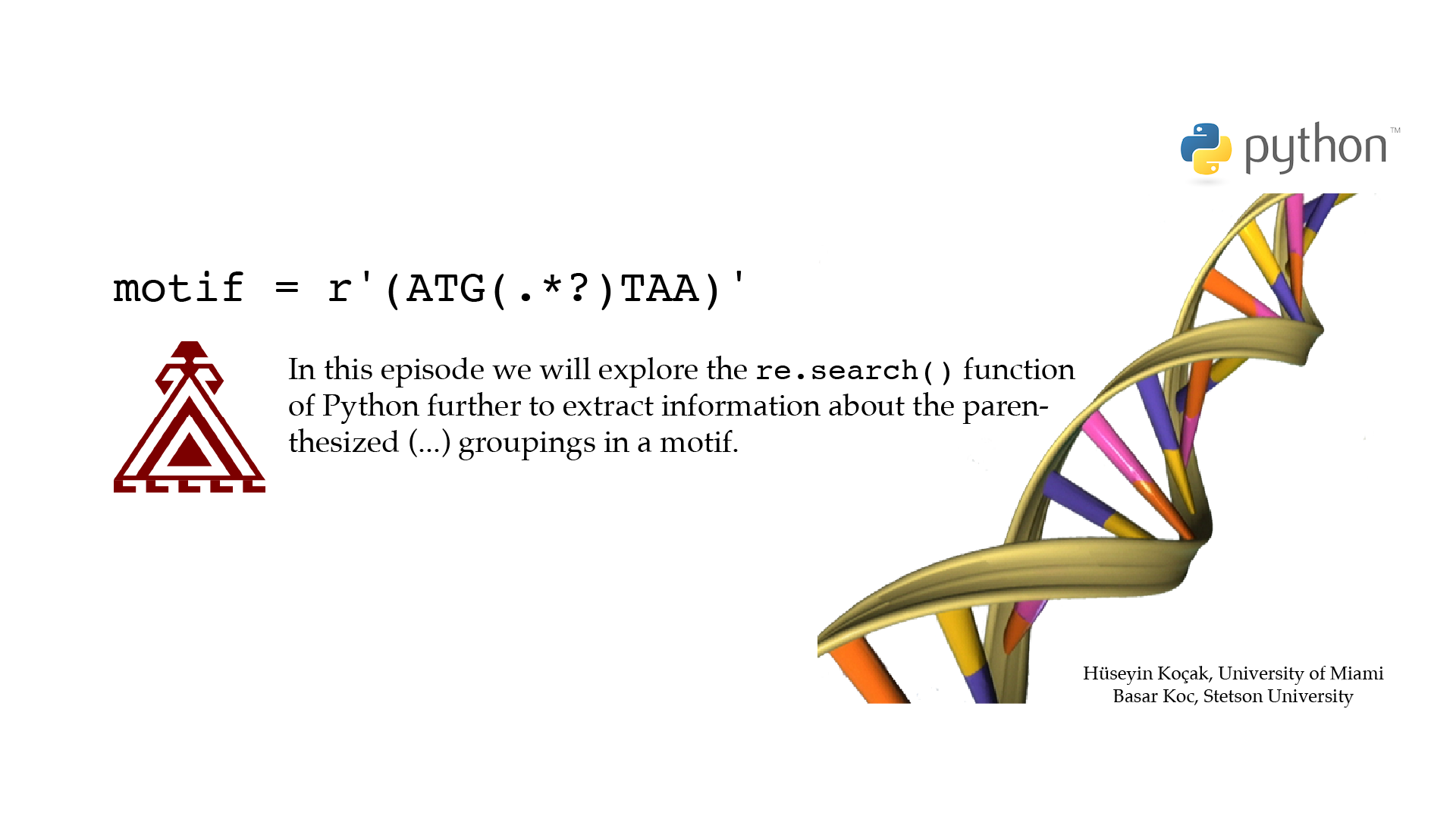
Motif Find All
# ------------------------------------------------------------------ # File name: motif_findall.py # # re.findall(pattern, string, flags=0) # Return all non-overlapping matches of pattern in string, as a list # of strings. The string is scanned left-to-right, and matches are # returned in the order found. If one or more groups are present in # the pattern, return a list of groups; this will be a list of tuples # if the pattern has more than one group. Empty matches are included # in the result. # # This program finds all the occurrences of a motif # in a DNA sequence and reports the motifs found as a list. # The motif, in regular expression, here consists of # substrings of A and/or T of lengths between 3 and 6, with two groups. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/re.html#re.findall # https://docs.python.org/3/library/re.html#module-re # https://docs.python.org/3/library/re.html#raw-string-notation # https://docs.python.org/3/library/re.html#re.compile # http://www.ncbi.nlm.nih.gov/nuccore/XM_002295694.2?report=fasta # ------------------------------------------------------------------ import re import sys DNA_sequence = 'AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG' print('DNA_sequence:', DNA_sequence) motif = r'(([AT]){3,6})' print('Motif:', motif) # Checking if motif is a valid regular expression try: re.compile(motif) except: print('Invalid regular expression, exiting the program!') sys.exit() matches = re.findall(motif, DNA_sequence) if matches: print('List of matches:', matches) else: print('Did not find any match.')[csc210@boston myPython]$ python3 motif_findall.py DNA_sequence: AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG Motif: (([AT]){3,6}) List of matches: [('AAT', 'T'), ('ATAA', 'A'), ('TAATTT', 'T')]
Download

Motif Find Iter
# ------------------------------------------------------------------ # File name: motif_finditer.py # # re.finditer(pattern, string, flags=0) # Return an iterator yielding match objects over all non-overlapping # matches for the re pattern in string. The string is scanned # left-to-right, and matches are returned in the order found. # Empty matches are included in the result. # # The program herein finds all the occurrences of a motif # in a DNA sequence and reports the motifs found as a match object # which contains position information. # The motif, in regular expression, here consists of # substrings of A and/or T of lengths between 3 and 6, with one group. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/re.html#re.finditer # https://docs.python.org/3/library/re.html#finding-all-adverbs # https://docs.python.org/3/library/functions.html#any # https://docs.python.org/3/library/re.html#module-re # https://docs.python.org/3/library/re.html#raw-string-notation # https://docs.python.org/3/library/re.html#re.compile # http://www.ncbi.nlm.nih.gov/nuccore/XM_002295694.2?report=fasta # ------------------------------------------------------------------ import re import sys DNA_sequence = 'AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG' print('DNA_sequence:', DNA_sequence) motif = r'([AT]){3,6}' print('Motif:', motif) # Checking if motif is a valid regular expression try: re.compile(motif) except: print('Invalid regular expression, exiting the program!') sys.exit() motif_found = False matches = re.finditer(motif, DNA_sequence) for match in matches: print('-'*20) print('%02d-%02d: %s' % (match.start(0), match.end(0), match.group(0))) print('%02d-%02d: %s' % (match.start(1), match.end(1), match.group(1))) motif_found = True if motif_found == False: print('Did not find any motif match!')[csc210@boston myPython]$ python3 motif_finditer.py DNA_sequence: AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG Motif: ([AT]){3,6} -------------------- 00-03: AAT 02-03: T -------------------- 17-21: ATAA 20-21: A -------------------- 30-36: TAATTT 35-36: T
Download

Motif Search Interactive
# ------------------------------------------------------------------ # File name: motif_search_interactive.py # # re.finditer(pattern, string, flags=0) # Return an iterator yielding match objects over all non-overlapping # matches for the re pattern in string. The string is scanned # left-to-right, and matches are returned in the order found. # Empty matches are included in the result. # # The program herein stores a DNA sequence in uppercase letters and # asks the user to enter a motif in regular expression. If the motif # is an invalid regular expression, it asks the user to enter another # motif. If the regular expression is valid, it finds all matches of # the motif in the DNA sequence and reports the matches, including # groups and positions. Matches are displayed in lowercase letters. # If no motif is entered, the program terminates. # # Version: 2.1 # Authors: H. Kocak and B. Koc # University of Miami and Stetson University # References: # https://docs.python.org/3/library/re.html#re.finditer # https://docs.python.org/3/library/re.html#module-re # https://docs.python.org/3/library/re.html#raw-string-notation # https://docs.python.org/3/library/re.html#re.compile # http://www.ncbi.nlm.nih.gov/nuccore/XM_002295694.2?report=fasta # ------------------------------------------------------------------ import re DNA_sequence = 'AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG' print('DNA_sequence:', DNA_sequence) while True: motif = input('Enter a motif to search for or enter to exit : ') if motif == '': break print('Motif:', motif) print('-'*len(DNA_sequence)) # Checking if motif is a valid regular expression try: re.compile(motif) except: print('Invalid regular expression!') else: motif_found = False matches = re.finditer(motif, DNA_sequence) for match in matches: for i in range(0, len(match.groups()) + 1): print('group%02d %02d-%02d: %s' % (i, match.start(i), match.end(i), match.group(i))) DNA_sequence = DNA_sequence[0:match.start()] + match.group().lower() + DNA_sequence[match.end():] print('-'*len(DNA_sequence)) motif_found = True if motif_found == True: print('Motif search is completed:') print(DNA_sequence) DNA_sequence = DNA_sequence.upper() print(DNA_sequence) else: print('Did not find any motif match!') print('~'*len(DNA_sequence)) print('Bye!')[csc210@boston myPython]$ python3 motif_finditer.py DNA_sequence: AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG Enter a motif to search for or enter to exit : ([AT]){3,6} Motif: ([AT]){3,6} --------------------------------------- group00 00-03: AAT group01 02-03: T --------------------------------------- group00 17-21: ATAA group01 20-21: A --------------------------------------- group00 30-36: TAATTT group01 35-36: T --------------------------------------- Motif search is completed: aatGAAGGGCCGCTACGataaGGAACTTCGtaatttCAG AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ Enter a motif to search for or enter to exit : (([AT]){3,6}) Motif: (([AT]){3,6}) --------------------------------------- group00 00-03: AAT group01 00-03: AAT group02 02-03: T --------------------------------------- group00 17-21: ATAA group01 17-21: ATAA group02 20-21: A --------------------------------------- group00 30-36: TAATTT group01 30-36: TAATTT group02 35-36: T --------------------------------------- Motif search is completed: aatGAAGGGCCGCTACGataaGGAACTTCGtaatttCAG AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ Enter a motif to search for or enter to exit : ((.)(.)\3\2) Motif: ((.)(.)\3\2) --------------------------------------- group00 03-07: GAAG group01 03-07: GAAG group02 03-04: G group03 04-05: A --------------------------------------- group00 08-12: GCCG group01 08-12: GCCG group02 08-09: G group03 09-10: C --------------------------------------- group00 20-24: AGGA group01 20-24: AGGA group02 20-21: A group03 21-22: G --------------------------------------- group00 25-29: CTTC group01 25-29: CTTC group02 25-26: C group03 26-27: T --------------------------------------- group00 30-34: TAAT group01 30-34: TAAT group02 30-31: T group03 31-32: A --------------------------------------- Motif search is completed: AATgaagGgccgCTACGATAaggaActtcGtaatTTCAG AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ Enter a motif to search for or enter to exit : \1 Motif: \1 --------------------------------------- Invalid regular expression! ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ Enter a motif to search for or enter to exit : Bye!
Download

-
Deciphering regular expressions: Give an English translation and an example of each of the following regular expressions:
AT+C [AT]+ [^AT]+ (.)(.).\2\1 ((.)(.).\3\2) ^ACTA{3,}TTT$ -
Translating English to regular expressions: Translate the following English sentences describing motifs to regular expressions:
1. match TATA or nothing (Answer: (TATA)?) 2. match TATAA followed by A or T (such motifs are called TATA boxes) 3. match 3-mer consisting of the dimer CG that is not immediately followed by C or G 4. match and capture a palindromic 5-mer
- Is it alive? Write a Python code to test if a given sequence (string) is a DNA sequence (consisting of A,C,G,T only). You can test your code on the SARS-CoV-2 sequence (MT233521.1) with gaps. Do the same for a protein sequence.
-
Substitution with regular expressions:
We saw in the episode Replace how to use the function str.replace() to replace all occurances
of a substring in a string. The function
re.subn() performs a similar function but allows the substring
to be specified with regular expressions.
What does the Python code below do?
import re dna = "601 accacac gacgag gtattag" dna1 = re.sub(r"[\d\s]", "", dna) print(dna1) dna2 = re.subn('a', 'a', dna1) print(dna2) print(dna2[1]) -
Searching for patterns:
Create a Python program that can check whether a given DNA sequence
contains the pattern
TATAxxxATGxxxT
where xxx can be any three consecutive DNA bases. That means your program, given some string, should identify if TATA, followed by any three DNA base pairs, followed by ATG, followed by any three DNA base pairs, followed by T can be found in the given string. Check both the forward and reverse complementary strand (reverse complement). Create three examples, one with a DNA string that contains the pattern in the forward strand, one that contains the pattern in the reverse complement strand and one that does not contain it in either strand, to demonstrate how the program works. Please include print statements that identify each different case and the position of the pattern in the string, if found. -
Overlapping patterns:
Write a Python program that can print the number of times a substring with five
characters that starts with 'AT' and ends with 'A' appears in a given string.
This means that you should recognize the appearance of every pattern 'ATxxA',
where 'x' can be any character. Test your program with the following string:
DNA = ATAGACATATAGATATGTGCACATAGAGAGATATACACATATATGTACATACA
Hint: Use the following look-aheadre.finditer(r'(?=(AT..A))', DNA)
Answer: The number of times that the aforementioned pattern appears in the string above is 9.
-
Binding sites in mRNA:
Shine-Dalgarno sequence is a binding site in the mRNA, usually located 8
base pairs upstream of the start codon AUG. The concensus sequence is AGGAGG. Create
a program that can examine any given FASTA sequence for occurences of
the Shine-Dalgarno sequence upstream of a start codon.
You will have to examine both the forward and complementary strands.
You have to report
the occurence of the AGGAGG pattern before any ATG, with 0-20
bases separating them.
Report separately the number of occurences for each length value of the segment between the two sequences. For example, you should report the number of times AGGAGG appears directly before ATG, the number of times AGGAGG appears one base before ATG, etc., up to 20 bases between them.
Now test your code with the genomes of two bacterial chromosomes, both larger than 5MB, one from a gram-negative bacterium and another from a gram-positive bacterium. For a gram negative, you could download the genome of Pseudomonas Aeruginosa, where for gram positive you could use Desulfitobacterium hafniense, or select other genomes of your choice.
- Simulating scanf(): Python does not currently have an equivalent to scanf(). Consult here for some more-or-less equivalent mappings between scanf() format tokens and regular expressions. Now, modify input.py code to read in two numbers entered on one line, then print out their sum.
- Logistic growth model:
Consider the discrete logistic model
x[n+1] = r * x[n] * (1 - x[n])
modeling the growth of a single population, where x[n] is the density of the population at n-th generation and r is the growth rate. Fix the growth rate r = 3.1 and initial population density x[0] = 0.43. Write a Python program that computes the population densities for 20 generations and puts them in a list. Then, print out each generation and its population density on a separate line for all 20 generations. - FASTA to GenBank: Write a Python function that takes as a parameter the name of a FASTA file and writes out the content of the file in GenBank format.
- TATA-Pribnow box: Motifs TATAAA or TATAAT of six nucleotides, known as TATA boxes, are an essential part of a promoter site on DNA for transcription to occur. Write a Python program that finds all the TATA boxes in a DNA sequence and reports their types and locations. If there is no such a box, your program should report that as well. Test your program on several DNA sequences with none, and with several TATA boxes of both types.
- TATA boxes in ebola virus genome: Download the FASTA file (accession number KC545393) containing the ebola virus genome. (You can use the "Send" widget on the upper-right corner of the NCBI Web page containing the genome to download a FASTA file.) Now, modify your program from the previous problem to produce the same TATA box report of the ebola virus genome.
-
Palindromic restriction sites in Zika virus genome:
Restriction enzymes are enzymes that cut double-stranded DNA at specific recognition nucleotide sequences known as
restriction sites. Quite often, these sites are palindromic, which for double-stranded DNA means they read the same
on the reverse strand as they do on the forward strand when both are read in the same orientation. For example, the
following is a palindromic restriction site:
5'-GTATAC-3' |||||| 3'-CATATG-5'
Create a program that, given a DNA sequence, will output all palindromic DNA sites of length 6 and their location. Test your program with:
- The sequence: "GCTAGTGTATGCATGAGCGTAGGCGA TGTGGCGCCGAGCTGAGGTGATCACGTGATGTGCTAGTCG".
- Zika virus genome: Download the FASTA file (NC_012532.1) containing the Zika virus genome. (You can use the "Send" widget on the upper-right corner of the NCBI Web page containing the genome to download a FASTA file.)
| Special characters | Meaning |
|---|---|
| . | any character, except newline |
| \n | newline |
| \t | tab |
| \s | any whitespace character (space, newline, tab) |
| \S | any non-whitespace character |
| \d | any digit character |
| \D | any non-digit character |
| \w | any one word character (alphanumeric plus _) |
| \W | any one nonword character |
| * | match 0 or more times preceding character or group |
| + | match 1 or more times preceding character or group |
| ? | match 0 or 1 time |
| ? | shortest (non-greedy) match |
| { } | repetition |
| { , } | repetition, min to max |
| ( ) | capture |
| ( ) | grouping |
| \1 | recall the first capture |
| \2 | recall the second capture |
| \n | recall the n-th capture |
| ^ | beginning of string |
| $ | end of string |
| [ ] | any one of set of characters |
| [^ ] | except any one of set of characters |
| | | alternative |
| \ | special or quote |
| (?=...) | Positive look-ahead. Matches if ... matches next, but doesn’t consume any of the string |
| (?!...) | Negative look-ahead. Matches if ... doesn’t match next |
| Regular Expression | Example | Meaning |
|---|---|---|
| AGA | TAGATC | match AGA anywhere |
| ^AGA | AGATGC | AGA at the beginning |
| TAA$ | AAGTAA | TAA at the end |
| A.T | AAACTG | A followed by any single character (except newline), followed by T |
| A.*T | CATATCT | A followed by any number of characters, followed by T (greedy) |
| A.*?T | CATATCT | A followed by any number of characters, followed by T (non-greedy) |
| (A.*?T) | CATATCT | capture A followed by any number of characters, followed by T (non-greedy) |
| A{5} | TAAAAATC | 5 A's |
| TA{2,4}CG | CTAAACGA | T followed by 2 to 4 A's, followed by CG |
| [AT]CG | CCTTCGA | either of ACG or TCG |
| (AA|CC|TT)CG | ACCCGTA | AA or CC or TT, followed by CG |
| A(CG){3}T | GACGCGCGTA | A followed by 3 CG's, followed by T |
| ((.)(.)\3\2) | GAATTAC | capture 4 consecutive characters, 1st and 4th, and 2nd and 3rd the same |
| A\.T | TCGA.TAA | A followed by . and T |
| AAA(?=TAG|TGA|TAA) | TAAATGAT | AAA if followed by TAG or TGA or TAA |
- Useful links:
- Regular Expressions HOWTO
A gentle introductory tutorial to using regular expressions in Python with the re module. - re module of Python for Regular Expressions.
- Useful links:
- The Python Programming Language
This is the official Web site for Python. You can download the Python interpreter and the extensive standard library are freely for all major platforms from this site. - The style guide to Python programming
The guidelines provided here are intended to improve the readability of code and make it consistent across the wide spectrum of Python code. - The official Python Tutorial
This tutorial introduces many of Python’s most noteworthy features, and will give you a good idea of the language’s flavor and style. - Regular expressions summary with examples
- Biopython
Biopython is a set of freely available tools for biological computation written in Python by an international team of developers. - Perl for Biologists
A mirror image of the current website in the Perl programming language. - Perl for Exploring DNA, by Mark D. LeBlanc and Betsey Dexter Dyer, Oxford University Press, 2007.
- Beginning Perl for Bioinformatics, by James Tisdall, O'Reilly Press, 2001.
- NCBI Genome site
- NCBI Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome (SARS-CoV-2) (NC_045512.2)
- NCBI SARS-CoV-2 (Severe acute respiratory syndrome coronavirus 2) sequences from NIH GenBank.
- Latest research information on coronavirus from NIH
- NCBI Zika virus, complete genome (NC_012532.1)
- NCBI Bundibugyo ebolavirus isolate EboBund-112 2012, complete genome (KC545393.1)
- NCBI Thalassiosira pseudonana CCMP1335 chromosome 7 breast cancer 2 early onset (BRAC2) mRNA, partial cds (XM_002295694.2)
- Pan troglodytes verus isolate MABEL mitochondrial D-loop (Chimp (AF176731.1)
- H.sapiens mitochondrial DNA for D-loop (Human) (X90314.1) and
- The Entertainer, by Scott Joplin.